X-ray crystallography
- Jay Sardesai

- Dec 17, 2021
- 4 min read
X-ray crystallography is the predominant method used today to find the structure of crystals and large molecules such as protein. X-ray crystallography works by directing a beam of light towards the crystal, and detecting the x-rays which diffract around the atoms. The structure can then be deduced from the intensity of the light detected at different angles.
X-ray crystallography relies on a process called diffraction. Diffraction is the spreading out of a wave as it passes near an obstacle, or through a slit. Usually, the rate of diffraction depends on the ratio of the wavelength to the size of the slit. The smaller that the slit is relative to the wavelength, the greater the diffraction. In this case, however, a different process occurs. When the X-rays hit an atom, they are scattered, due to its electrons, and form secondary circular waves around the atom.
Using Bragg's law (2dsinθ =nλ), where d is the distance between each "sheet" of atoms in the crystal, θ is the angle of incidence (where the plane of the sheet is considered to be a mirror), and nλ is the path difference. Path difference is the difference, at a point, assuming that the waves have the same wavelength, of the number of wavelengths it took for the waves to move from the source to the point. The points at which the path difference is an integer can be found as constructive interference occurs there, instead of destructive interference, leading to a peak on the diffractogram, called a Bragg peak. Also, the sources are considered to be the atoms which scattered the X-rays, not the original source of the X-rays. Using Bragg's law, the dimensions of the unit cell can be calculated, however, the arrangements of the atoms inside the crystal is still not known, and therefore a Fourier transform is needed, where a complex wavefunction is separated into its constituent waves, allowing the precise arrangement of the atoms to be deduced.
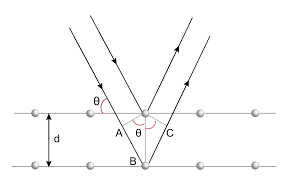
To find the structure of a crystal, a monochromatic beam of X-rays is directed at it, and the crystal is slowly rotated, changing the interference pattern observed. Using the interference pattern, the points of scattering can be found, allowing the position of the electrons to be deduced. From this, the structure of the crystal can be found. There are, however, some limitations to this method; as the number of atoms in the unit cell increases, the resolution of the image obtained decreases. In addition, it's usually much easier to create crystals if the repeating unit is smaller.
The crystals used for this method must be very regular, and by extension pure. Otherwise, the structure deduced will be flawed and inaccurate. For this reason, the crystals used are often synthesised in the lab. The most important area in which this is used is in the analysis of proteins, as unless they are crystallised, the diffractogram produced is very blurred and potentially useless. Large biological complexes can also be crystallised, like viruses. This mechanism was first observed by Friedrich Hünefeld in 1840, when he found crystallised haemoglobin in earthworms. Since then, it has been used to find the structure of countless proteins and has been an invaluable tool in the field of biochemistry.
To induce protein crystallisation, first, the protein must be purified to as close to 100% purity as possible. Then, it must be concentrated to the highest possible concentration which does not cause precipitation of the protein. Afterwards, a precipitating agent can be introduced which causes precipitation of the protein in the form of crystals. One method in which this can be achieved is with vapour diffusion, where the protein solution is placed as a hanging drop above a much more highly concentrated solution of the protein and precipitating agent. Over time, the concentration of the protein increases in the drop by diffusion, and under the right circumstances, precipitates to form a crystal. The formation of the crystal can be encouraged by optimising some of the conditions, such as pH and temperature, of which the ideal is different depending on the protein.
The field of X-ray crystallography was made possible by Max von Laue who, in 1912, discovered X-ray diffraction. In addition to this, he created the Laue equations: Δk = 2πh, Δk = 2πk, and Δk = 2πl, where Δk is the difference between the outgoing and incoming wavevectors, and h, k and l are the Miller indices used to denote planes in crystal lattices. The Laue equations are used to predict the paths of X-rays through a lattice where the structure of the lattice is known; so they can also be used to deduce the structure of the lattice from the path of the waves.
X-ray crystallography can also be used to find the structure of catalysts, which can help determine their viability. In addition, by observing the structure of a catalyst as its temperature changes, its stability under stress from heat can be found, in order to see whether it is suitable for an application at a certain temperature
X-ray crystallography has been involved in many very important discoveries. In 1913, researchers used it to confirm the theorised structure of diamond (a tetrahedral arrangement of carbon atoms). Perhaps more importantly, in 1952, Rosalind Franklin used X-ray crystallography to create an image of DNA, which allowed Watson and Crick to deduce its structure. Unfortunately, she died in 1956 of ovarian cancer, possibly related to her exposure to X-rays, and thus was not eligible for the Nobel Prize, which Watson, Crick, and Watkins shared in 1962.
Overall, X-ray crystallography has proved itself to be an invaluable tool for modern science, with 14 separate Nobel Prize-winning discoveries since 1914 made possible by X-ray crystallography. Without it, it is likely that many of these discoveries would not have been made, and for that reason X-ray crystallography is of pivotal importance to modern science.
Sources:


Comments